| |
| DNA = contingent |
YES |
because there are no restrictions for DNA-base combinations: any sequence of DNA-bases can exist. The sequence of bases in DNA is not derivable from the chemical properties of the bases. It is the freedom of the sequence that DNA and language have in common. |
| DNA = complex |
YES |
because millions of bases do occur in organisms and they are not repetitions of simple sequences. Although repetitious DNA exists and genes are duplicated, organisms harbour thousands of genes and each gene has a unique sequence of hundreds of bases. |
| DNA = information |
YES |
because it is a linear sequence of contingent symbols, the 4 bases. |
| ? | but see paragraph Paradoxes & Limitations. |
| DNA = specified |
? |
Dembski did not give a definition of 'specified DNA', so we cannot establish if a particular DNA sequence is 'specified'. [see below] |
| DNA = designed |
YES |
if 'designed' is used as a metaphor and means: 'has a function'. In that case it adds nothing to the concept 'specified'. |
| NO |
"designed DNA" is not a useful scientific concept if it implies that the origin of "designed DNA" is not accessible to science. |
| DNA = intelligently designed |
YES |
if that means 'optimally designed for its task', although we simply don't know this for most genes. And Yes, if 'intelligent' means 'choosing between' ! (p144), because natural selection chooses between genetically different individuals by differential reproduction. So according to Dembski's definition natural selection is intelligent. |
| ? |
because we don't know what 'intelligently' means if there is no method to measure 'intelligent' apart from 'designed'. According to Dembski's definition a rat in a maze is as intelligent as the Intelligent Designer designing a universe? |
|
|
|
Complex Specified Information of more than 500 bits is designed
Dembski defines Complex Specified Information, CSI, as "any specified information whose complexity exceeds 500 bits of information" (p166). Chance cannot generate CSI. However he didn't translate that into a concrete DNA sequence length. I did not find it in his book [20]. If he consulted Yockey [11] he would have noticed that the cytochrome-c family of genes (113 amino acids, 339 bases long) has an information content of 233 - 373 bits. This is clearly lower than 500 bits. So although cytochrome-c undoubtedly contains information, it does not classify as CSI. So cytochrome-c is not an example of design and could be generated by chance according to Dembski's criterion. This is a rather unexpected result. I don't doubt Yockey's calculation. Yockey made the most detailed calculation that can be found in the literature. Dembski knows Yockey's book. It has far reaching consequences for Dembski's design concept that cytochrome-c is not CSI. Because many genes have the same information content as cytochrome-c and many genes
are below the 500 bits boundary. Thus many genes would not classify as CSI and would not be designed. Histone-H3 is just above the boundary (505 bits). The largest known mammalian gene, the dystrophin gene [16], contains 79 pieces (exons), is 2.4 Mb (Million bases) long, encodes a protein of 3685 amino acids and if the sequence is specified, it certainly must be designed. However Dembski did not mention any gene by name. The only concrete claim Dembski has made is: "the CSI of a flagellum far exceeds 500 bits" (p178),
but there are about 50 genes involved in a flagellum. Why not claim that the human genome (estimated size: 3,2 billion base pairs; estimated number of genes: 30,000 - 40,000 25)
far exceeds 500 bits? Why stop there? If there are now a standing diversity of 100 million biological species and each had a hundred thousand genes and genes in each species were at least slightly different from genes in all other species, then, not counting molecular diversity within species, the number of genes on planet Earth is 10 trillion [18]. Dembski's problem is: what units do we use? a piece of DNA? an exon? a gene? a gene family? a functional group of genes? a whole genome? all genomes on Earth?
But there is a far more serious problem: it is easy to find genes with more than 500 bits of information. However only specified DNA sequences are to be considered according to Dembski's definition of CSI. So: What is 'specified' DNA?
|
|
| |
What is a 'specified' DNA sequence?
We need to distinguish between known genes and DNA with an unknown function. How do we know if a random piece of DNA is a gene? Well, if we know the protein it produces, it seems easy. If we don't know the protein, we could look in other organisms
for exactly the same sequence of DNA. If we find an exact match the sequence is probably not a random sequence but a gene. As we will see this is far from finding a 'specification'.
An example of a known gene is hemoglobin. The hemoglobin of the llama differs from that of the closely related camel by a single mutation, which confers on it a greater affinity for oxygen [1]. This is nice because the llama lives at high altitudes. There is a point mutation of hemoglobin called the "sickle-cell mutation" where one Thymine is
changed into one Adenine, resulting in sickle-cell disease. In both cases there isn't a huge informational barrier between the forms of hemoglobin. In fact it is the smallest possible difference of Information Content. Now, what is the "true" target or specification of the hemoglobin sequence? Is the sickle-cell form of hemoglobin 'un-specified'? Is one random mutation the difference between 'specified' and 'unspecified' information?
Another example is the target sequence of enzyme cytochrome-P450. Artificially created cytochrome P450 is up to 20 times more efficient than the natural cytochrome-P450 [2]. So what is the "true" target sequence: the natural or the artificial? Probably a whole range of sequences depending on the organism and its environment. If the target
is big, it is easy to hit. If it is small, it's difficult to hit. The conclusion is that even when we know the proteins, we cannot give one
'correct specification' for it.
The situation is worse than that. According to Dembski's own criteria a specified pattern needs to be independent of the event. The event is the observed DNA sequence. So we cannot use the observed DNA sequence to construct the specification for that DNA sequence. The specification must be constructed without recourse to the event. A typical example of a specification is the Fibonacci algorithm that generates an endless row of numbers, but the algorithm itself is very short (see paragraph below). This kind of algorithm can definitely not generate the information in DNA! It seems that neither a mathematical nor a non-mathematical specification of DNA is possible. It is extremely difficult to specify in advance which genes an organism needs, let alone to specify the precise sequence of bases. To know the sequence is one thing (biologists completed the task for the human genome), but to specify in advance independent of the observation what a sequence must be, is beyond current biological knowledge. The following remark of Dembski makes me even more pessimistic: "A random ink-blot is unspecified; a message written with ink on paper is specified. The exact message recorded may not be specified, but orthographic, syntactic, and semantic constraints will nonetheless specify it." [20,p189].
Law of Conservation of Information
Even if we assume that DNA contains Complex Specified Information (CSI),
this does not answer the question: what real-life mechanism could produce CSI?
Dembski concludes that natural law plus chance
cannot explain information content in DNA. Is that true?
To find out we must distinguish between two questions:
- is there a natural mechanism that creates the very first information (= origin of life)?
- can natural selection and mutation increase information content of DNA?
Biologists don't know all the details of the solution of the first question: the origin of life. The simplest free-living organism, Mycoplasma genitalium, has 468 genes. This would exceed Dembski's boundary of 500 bits, I guess. Could this evolve gradually? We need data and experiments.
Now the second question. Although Dembski tries to escape a positive answer to the second question, he finds himself saying: "selection introduces new information" (p177). Dembski also seems to accept that information can flow from the environment to an organism, thereby increasing the organism's information content. Both statements contradict his main thesis that natural processes cannot generate CSI. On other pages he is so attached to the Law of Conservation of Information ('Only Information begets Information', p. 183) and the belief that CSI cannot be generated by natural processes, that he is forced to believe that CSI existed before the origin of life: CSI could be 'abundant in the universe' and 'CSI is inherent in a lifeless universe'. This amounts to free-floating ghostly information in space, which is too far removed from down-to-earth biological science. The whole idea that information in DNA has any meaning outside living organisms is caused by pushing the information metaphor too far. The information in DNA is meaningless outside the cell. Just like the instructions in software do not have any meaning outside the very specific hardware environment in which they are executed. Further Dembski believes in 'discrete insertions of CSI over time in organisms' (p171). In that case I prefer Fred Hoyle's panspermia theory, which is as unearthly but closer to observational science.
Flow versus Origin
Dembski makes a useful distinction between the flow and the origin of
information. When information flows from one organism to another no information is generated and no special explanation is needed. However the origin of information needs an explanation. I agree with the distinction. Evolutionary biologists need a mechanism that generates information, because there was no CSI available on the Earth 4 billion years ago. According to Dembski Darwinists often claim to have explained the origin of genetic information, while in fact they only have explained the flow of information. The main thesis of Dembski's book is that an intelligent designer is a valid explanation for the origin of Complex Specified Information. But how can he accuse Darwinists of only explaining the flow of information, while his own explanation of CSI relies on pre-existing CSI? For Dembski holds that "To explain an instance of CSI requires at least as much CSI as we started with."
But then we have a case of the flow of information: pre-existing information flows from the designer to an organism. And then a designer doesn't explain the origin or the creation of information. Dembski virtually defines CSI as unexplainable
[14]. But then he did nothing to explain the origin of information.
Paul Davies
To understand why Dembski's design inference is in trouble let's have a look at Paul Davies' [3] description of the information content of DNA. Paul Davies describes 'contingent' as random strings and 'Complex Specified' strings as a subset of random strings. The subset cannot mathematically be defined, because "all random sequences of the same length encode about the same amount of information" ([3], p. 119). But that means that from an information-theoretical point of view all genes of the same length have the same information content! Furthermore they also have the same information content as an arbitrary random DNA sequence of the same length! So information theory can be applied to DNA, but it is powerless precisely where Dembski needs it most. Davies concludes that DNA sequences can only qualitatively (in a non-mathematically way) be subdivided. The biologically relevant subset can only be defined in
biological terms. Dembski needs the concept 'specified DNA', but how to define it without knowledge of the target DNA? The target DNA-sequences can only be discovered (if ever) by biologists experimentally, not calculated by mathematicians.
I think Dembski must agree with Davies' claim: "Random mutations plus natural selection are one surefire way to generate biological information, extending a short random genome over time into a long random genome." [3, p120]. Dembski must accept that genes like cytochrome-c can be produced by mutation and natural selection, because they are smaller than 500 bits.
If selection can do this: what are the limits? Can humans evolve from first life in this way? Just like other IDT, Dembski accepts that natural selection can produce 'micro-evolution', but not macro-evolution. This has implications for Dembski's Law of Conservation of Information. It is difficult to see how 'micro-evolution' could occur without increase in the information content of all the species involved. Again: what are the limits? Dembski did not calculate where the micro/macro borderline is. Without that borderline, the objection to the micro-to-macro extrapolation is un-mathematically vague.
We don't know yet how life originated, so we cannot say how the first information was created. However a first clue to the solution could be the fact that DNA sequences are mathematically described as contingent or random! Stuart Kauffman's autocatalysis model is based on random catalysis.
Kauffman's complexity theory
Stuart Kauffman's Autocatalytic Set theory is a theory about the origin of life and by implication of information. Therefore it is a direct competitor of Dembksi's Intelligent Design theory. It is ultimately based on a natural law plus chance. Thus one would expect that Dembski devoted time and space to refute Kauffman's theory. Furthermore both have a mathematical theory of information, both theories could be labelled 'complexity theories' and both men are mathematicians. So Dembski must be able to give a detailed evaluation of Kauffman's theories. Nothing of the sort happens in Intelligent Design. His criticism of the Mandelbrot fractals of the Complexity theorists of the Santa Fe Institute (p162-165) is interesting and very useful, but irrelevant as a criticism of Kauffman's autocatalytic set theory. Remarkably in the Appendix of his book he defends the relevance of mathematics to biology: "Mathematics does indeed elucidate biological complexity" (p271). He even goes so far to defend anti-reductionism which is inherent in "complex systems theory" of the Santa Fe Institute (where Kauffman is working):"Complex systems theory has long since rejected a reductive bottom-up approach to complex
systems" (p256).
Elsewhere [4] Dembski wrote a short and hostile review of Kauffman's work, claiming that there are neither laws of self-organisation nor biological evidence for complexity theory. It is remarkable that one mathematician criticises the other mathematician for lack of real-life evidence, but of course both Kauffman and Dembski need to demonstrate the relevance of their theories for real-life. Kauffman needs evidence as much as Dembski needs to point out that his definition of CSI is relevant to problems in evolutionary biology (see below). No scientist can practice science by mathematics alone. Dembski's attitude towards Kauffman appears even more remarkable when one realises that Kauffman's line of research makes criticism possible of the omnipotence and creativity of natural selection.
Perakh's criticism of Dembski's design inference
 |

Which is designed? |
|
Physicist Mark Perakh asks a seemingly innocent and simple question: If we find pebbles and a perfect spherical white ball on the beach, which is designed?
Nobody needs advanced mathematics to decide which is designed and which originated by random forces. However, the information content of the spherical white ball can be described by a very short formula only containing its diameter and colour. Therefore, the algorithmic information content of the ball is low. Contrary, the algorithmic information content of an arbitrary pebble is very much higher because a very complex formula is required to describe its irregular surface and colour distribution. "This example again illustrates that complexity in itself is more likely to point to a spontaneous process of random events while simplicity (low complexity) more likely points to intelligent design. This is contrary to the definition of complexity given by Dembski." [24].
Paradoxes, Limitations, Open Questions, False Positive, Fine Tuning
Paradoxes
One of the paradoxical implications of the definition of Information Content of a string of symbols is that a random string contains the maximum amount of information [5]. Which is of course the opposite of how we use 'Information Content' in everyday life. So we must be extremely careful when applying the concept outside its mathematical context. "The ability to store information is not very interesting for evolution. You have to be able to transfer that information into a molecule that does something." [55]
Limitations
There are limitations of the mathematical concept of information. Above we saw already that all genes of the same length have the same amount of information (Davies) and the same amount of information as a random string! This shows clearly
the limitations of information theory applied to DNA. Clearly something is missing: meaning or quality. Dembski added 'specification', but did not define 'specified DNA'.
Furthermore the Information paradigm is an example of reductionism. For someone who believes that the 'possession' of information is the only thing that matters, it will be no surprise to see a woman with a moustache and beard, since we know that all humans possess the gene for the production of hair. And the information is present in all body cells. So the presence of information in DNA does not fully explain the living organism. What matters is gene regulation: the expression of information. Compare this with the static and linear information of a book: 100% of the information is 'expressed' from the first to the last page and is also designed to be read in that order. A human body is not build by reading its genetic information from chromosome 1 through 23. A computer program with complex behaviour is a better model than a static book. Kauffman captured just this dynamic aspect in his models: the behaviour of a system.
In a more general sense the software or information in a book analogy is misleading. Master copies of software and books ('the original') are kept in a safe, but the human genome does not have a master copy. The human genome is a copy of a copy of a copy, etc. There is no original.
| |
|
Open questions
The information content of 'the human genome' (the Human reference genome) is usually defined as the sequence of the 4 bases. However this definition ignores:
- Definition. How to define the information content of the genome? There is no such a thing as the information content of 'the human genome'. This is because there is genetic variation between individuals, and variation between the tissues and cells of the same individual. Further, each human being has two genomes: the body cells are diploid (n=2; 46 chromosomes). Furthermore, females have sex chromosomes XX and males have XY. Since the X chromosome is larger than the Y chromsome, females have more DNA than males. Additionally females don't have the Y-chromosome. And finally, Down syndrome patients have an extra chromosome 21 (they have 47 choromosomes), should that be included or ignored?
The remarks above suppose genetic information in DNA is like (static) information in a book. This is wrong. DNA is 'read' dynamically in a complex way to create and maintain a body. The information embedded in 'reading' the genome is a least just as important as the information in DNA itself. [added: 3 July 2020]. Much more trouble is listed below.
 Genetic variation (polymorphisms): The human genome was published in 2001, but the DNA sequence of any individual does not exactly match this reference sequence. Each individual genome contains millions of polymorphisms – variable sites that differ from the reference. "Every individual has tens of thousands of rare non-coding variants, which are often ignored in a clinical context and in disease studies." [48]. There are about 12.5 million DNA bases known to vary between individuals [48]. Three years later: "We identified 67.3 million single-nucleotide polymorphisms, 8.8 million small insertions or deletions (indels), and 40,736 copy number variants in 929 genomes from 54 geographically, linguistically, and culturally diverse human populations" [61]. In 2020 structural variants of 17,795 individuals were sequenced. On average, individuals carry 2.9 rare structural variants that alter coding regions; these variants affect the dosage or structure of 4.2 genes and account for 4.0–11.2% of rare high-impact coding alleles [64]. So, the reference genome is a theoretical construct, and so is "the information content of the human genome". Genetic variation (polymorphisms): The human genome was published in 2001, but the DNA sequence of any individual does not exactly match this reference sequence. Each individual genome contains millions of polymorphisms – variable sites that differ from the reference. "Every individual has tens of thousands of rare non-coding variants, which are often ignored in a clinical context and in disease studies." [48]. There are about 12.5 million DNA bases known to vary between individuals [48]. Three years later: "We identified 67.3 million single-nucleotide polymorphisms, 8.8 million small insertions or deletions (indels), and 40,736 copy number variants in 929 genomes from 54 geographically, linguistically, and culturally diverse human populations" [61]. In 2020 structural variants of 17,795 individuals were sequenced. On average, individuals carry 2.9 rare structural variants that alter coding regions; these variants affect the dosage or structure of 4.2 genes and account for 4.0–11.2% of rare high-impact coding alleles [64]. So, the reference genome is a theoretical construct, and so is "the information content of the human genome".
- Epi-genetics:
- Epi-genetics: DNA is nothing without its proteins. At any one time, tens of thousands of proteins are latching onto or backing away from the genome, creating the dynamic biochemistry that fuels life. The histone code hypothesis describes how histone modifications can convey information. (B. Turner, Nature Cell Biol. 9, 2 (2007)).
- Epi-genomics: the epigenome is a genome-wide map of reversible chemical modifications to DNA and its associated proteins that determine when genes can be expressed. "By 2004, large-scale genome projects were already indicating that genome sequences, within and across species, were too similar to be able to explain the diversity of life. It was instead clear that epigenetics could explain much about how these similar genetic codes are expressed uniquely in different cells, in different environmental conditions and at different times." "Epigenetic coding will be orders of magnitude more complex than genetic coding". (Nature 4 Feb 2010). "The human genome is singular and finite, but the human epigenome is almost infinite - the epigenome changes in different states and different tissues". Every cell in our body has its own epigenome, that is 1015 different epigenomes (Marianne Rots). That is a huge amount of information. There are 3 chemically modified versions of Cytosine and 1 methylated Adenine. Methylated Adenine plays a key role in the development of the placenta [65].
- Epi-transcriptome: all chemical modifications to mRNA (that is after transcription). Transcriptome is the complete set of mRNA molecules found in a cell. The fate of messenger RNAs is determined by the sequence of their four main molecular building blocks: A (adenosine), G (guanosine), C (cytidine) and U (uridine). However, these components can be chemically modified in ways that impart additional information to mRNAs. (Epitranscriptomics).
- Gene regulation or the regulatory code: "Numerous observations suggest that the amount of regulatory DNA associated with the average gene in a multicellular species is at least as great as the length of the coding region" (source). An organism's genome consists of a complex code that specifies not only the DNA sequence of genes but also how and when they are transcribed.
"We were surprised to find how much information hides in the combinatorial patterns of chromatin marks. Instead of simply ON or OFF information, we found that we could recognize different functional classes of genes solely based on their chromatin patterns." "There are over 100 ways that chromatin can be modified, and researchers have hypothesized that specific combinations of changes to chromatin may lead to different biological ends" (source).
Furthermore, there is X-chromosome inactivation in females. One of the two X-chromsomes in a body cell is randomly inactivated. This does not alter the sequence, but the expression of genes. The random nature adds information. Should that be ignored?
- one-gene-one-protein hypothesis is no longer valid; for example human genes produce on average 10 proteins.
So the real information content is 10 times the number of genes?
- proteome: the sum of human proteins exceeds the number of genes by far, and could run in the millions. Where and when are genes translated? When in the development process? Where in the body, in which cells and tissues? What is the difference between male and female? and in cancer and other diseases? In the body, protein abundance varies by as much as 1-million-fold. All this should be included in the definition of information content of the human genome-proteome. [40]
- The three main questions of the protein-folding problem [35].
- The physical folding code: How is the 3D native structure of a protein determined by the physicochemical properties that are encoded in its 1D amino-acid sequence?
- The folding mechanism: A polypeptide chain has an almost unfathomable number of possible conformations. How can proteins fold so fast? (seems proteins have a fast and efficient search algorithm?)
- Predicting protein structures using computers: Can we devise a computer algorithm to predict a protein's native structure from its amino acid sequence? (if succesful, these computer algorithm contain information not present in the dna sequence?)
We cannot consistently predict the structures of proteins to high accuracy from the sequence of the amino acids. Every amino acid can be coupled in 10 geometrical ways with its neighbour. So a sequence of 200 amino acids has 10200 possible configurations. This cannot (yet) be computed by any number of (super)computers [39]. The protein itself cannot try out all these configurations and yet protein folding is done in (milli)seconds. This is called the Levinthal paradox. Folding diseases: Alzheimer's, Parkinson's, and type II diabetes. End 2020 Google's AlphaFold software solved the problem [66].
- Two conformations: hemoglobin is the best-known example of a protein that can exist in two conformations,
two overall shapes of the same protein (with exactly the same amino acid sequence). One form is oxygen-loading and the other is the oxygen-unloading form (23). Dembski's definition of information content of the protein ignores this extra and highly functional kind of information, because his definition is based only on the linear sequence of amino acids in the protein or bases in DNA.
- Development of the embryo: A DNA sequence is a one-dimensional entity, and an embryo is an 3-D entity. Since there is no miniature baby in a fertilised egg, how does one get from 1-dimensional information in DNA to a 3-dimensional organism? There is a real increase in geometrical complexity during development [13]. Compare this again with the 1-dimensional information in a book. Is 1-dimensional information in DNA sufficient for building a 3-dimensional organism? [8]. The development of the embryo shows cell behavior that depends on the spatial position of the cell [49]. These spatial positions can be considered as new information which is absent from one-dimensional DNA. So, the total information content of an organism increases in the development from fertilised egg to adult. According to Dancoff & Quastler the information content of the germ plasm is 106, and of the adult roughly 1025. That increase would be enough to refute Dembski's Law of Conservation of Information.
- Increase of information for free:
Increase of genetic information for free:
- development of the embryo: "Every cell of your body was generated by cell division, forming a lineage tree that goes back to the fertilized egg. Mutations are introduced by errors in DNA replication at every cell division, as well as by mutational processes that operate continuously, such as exposure to ultraviolet light. As a consequence, every cell may have its own unique genome, with potentially distinct gains and losses of function." [45]. Genetic changes (mutations) in utero set the scene for Wilms' tumour, which is the most common childhood kidney cancer [59]. All cancers are baased on an increase of genetic information for free.
- children: When parents have children the total information content of DNA of the family increases in a natural way. If each child is genetically unique (except identical twins) then the information content of the family must increase. The reason is the 'Combinatorial explosion' of 23 out of 46 chromosomes of the father and 23 out of 46 chromosomes of the mother. The genomes of children cannot be computed from their parents genomes, if only because of unpredictable new mutations occur in sperm and egg cells. It violates Dembski's Law of Conservation of Information. See also: World population.
- Sex cells: males produce millions of sperm cells. Because of copying errors there will be also millions of differences in the exact DNA sequence. Mutations in egg cells also occur.
- trisomy-21 (Down syndrome): children with Down have 47 chromosomes. They have 1 extra chromosome 21. So, they have more genetic information than the rest of humankind.
- polyploidy is a process that produces a fourfold set (tetraploid) or more of the standard diploid chromosome set. After polypoidization event, mutations accumulate and the sets diverge. This increases the total information content of the genome. (wiki). Maize genome at one point [in evolutionary history] had four copies of each chromosome, but has evolved into an ostensibly diploid genome with a lot of duplication. Bread wheat is a hexaploid (2n = 6x7 = 42 chromosomes) (41). These are examples of increase of genetic information.
Polyploidy in liver cells: Many liver cells are polyploid, containing 4, 8, 16 (!) or more times the haploid chromosome complement, although the significance of the phenomenon is not known. This is somatic polyploidy. Polyploidy is a common characteristic of the mammalian hepatocytes. The resulting genetic heterogeneity [= increase of information!] might be advantageous following hepatic injury when 'genetically robust' cells could be selected from a pre-existing pool of diverse genotypes. (46)
- Cancer:
- cancer: increase of genetic information by random mutation in tumors. This is the status in 2013:
Recent studies have revealed extensive genetic diversity both between and within tumours. ... A major cause of genetic heterogeneity in cancer is genomic instability. This instability leads to an increased mutation rate and can shape the evolution of the cancer genome through a plethora of mechanisms. (37). The number of mutations in a cancer can vary from a handful (10–20) to (the more usual) hundreds or thousands. (38)
This is the status in 2018: a 2018 analysis of more than 9000 samples from cancer patients reported almost 1.5 million mutations (62). This is the status in 2020: 43,778,859 somatic single-nucleotide variations (SNVs), 410,123 somatic multinucleotide variants, 2,418,247 somatic small insertions and deletions (indels), 288,416 somatic SVs, 19,166 somatic retrotransposition events and 8,185 de novo mitochondrial DNA mutations. In total: 46,922,996 mutations, almost 47 million mutations. (63). Is this information destruction? Partly. Consider this: on the basis of the information in DNA normal somatic cells start behaving in dramatic new ways: cancer.
Neochromosomes: Above that, the genetic landscape of tumors is not static, but continually evolving. Cancer researchers introduced the word 'neochromosomes': Neochromosomes (in liposarcomas) are made up of pieces of the 46 chromosomes that each human cell normally carries. This heterogeneity is beneficial for the origin and evolution of tumor growth and the emergence of drug resistance. See also: [19].
By combining long-read DNA sequencing with RNA analysis, several previously undiscovered regions in which two genes were fused together, as well as nearly 20,000 alterations in the genome's structure were found. Many of these changes were missed by sequencing methods that analyse shorter fragments of DNA [51]. Please note that a new gene was created by random mutation. The new mutations are not by definition harmful or deadly for the cancer cells. It is thought to aid the cells' rapid adaptation to the effects of anticancer therapies (57).
- cancer: increase of genetic information: extrachromosomal circular DNA (ecDNA) was found in nearly half of human cancers; its frequency varied by tumour type, but it was almost never found in normal cells. Many cells with ecDNA have higher fitness and are more likely to pass these mutations on to daughter cells and proliferate. This is increase of information for free. (58)
- cancer: increase of protein information by random mutation in tumors: 56,592 different mutations in proteins in colon-cancer tissue from 110 people. Some of these had never been seen before (56).
- Chromothripsis: "is a mutational process by which up to thousands of clustered chromosomal rearrangements occur in a single event in localised and confined genomic regions in one or a few chromosomes, and is known to be involved in both cancer and congenital diseases." (Chromothripsis). Since it produces chromosomal rearrangements it creates new information for free. "Chromothripsis drives the evolution of gene amplification in cancer" [67].
- Aging. Increases in frequencies of mutations and chromosome rearrangements with increasing age, have been detected in a variety of cells and organisms. For example, DNA sequences called retrotransposons can copy themselves and reintegrate at new sites in the genome, causing damage [54] (and an increase of the total information content of the genome). Active retrotransposons ('jumping genes' ) are associated with genome instability during chronological aging. Somatic retrotransposition is a driver of aging (44).
All this increases the genetic information content of the aging body 'for free'.
- Alzheimer: Variable brain-specific mutations have been observed in Alzheimer's disease. One mechanism underlying this mosaicism involves integration of variant gene copies back into the neuronal genome by reverse transcription. This increases somatic genome and protein diversity in the brain. In total 6,299 different variants of the gene Amyloid-β Precursor Protein (APP) in the brains of 5 people with sporadic Alzheimer disease. In other words: increase of genetic information content. (52) This is an example of Somatic Genome Mosaicism.
- Somatic Genome Mosaicism: different cells in the body can have different mutations not present in the germ line, embryo and the rest of the body ('post-zygotic mutation'). For example: single-neuron genomics studies have identified remarkably diverse somatic mutations that reveal a wide gamut of mutation processes impacting the brain, from small point mutations and microsatellite polymorphisms to larger retrotransposon insertions, copy-number variants, and aneuploidy [47]. Cancers, which consist of genetically distinct cells, are a familiar example. This increases the genomic information content of the individual. [36], [43].
- HIV-1 (Aids virus):
1) Each time HIV infects a human immune cell, the virus copies its RNA genome into a DNA version that integrates at a new spot among the cell's chromosomes. So, the information content of the DNA of that cell increases (for free).
2)"Here is a stunning example of the consequences of RNA polymerase error rates. Tens of millions of humans are infected with HIV-1, and every infected person produces billions of viral genomes per day, each with one mutation. Over 1016 genomes are produced daily on the entire planet." (Virology blog). The information content of the whole virus population has increased.
- Viral integration in the human genome.
Viral Integration Site DataBase (VISDB) contains a total of 77.632 integration sites of five DNA viruses and four RNA retroviruses. Although viral DNA is existing DNA, for the human genome an integration of viral DNA in to the chromosomes is an increase of genetic information (60). The chromosomes become longer.
- World population: the information content of the genomes of the human population increases naturally because the world population increases steadily and humans are not clones. The total information content of all human genomes on earth is 6 exabyte (Hans Rosling, 28).
1 exabyte = 109 Gigabyte or 1018 bytes. Since Adam and Eve the infomation content of the human species has increased by something like a factor 109. (4 Apr 2011). Humans produce information in ever increasing quantities. "John Holdren, the president's science adviser, wasn't exaggerating when he said last week that "big data is indeed a big deal." About 1.2 zettabytes (1021) of electronic data are generated each year by everything from underground physics experiments and telescopes to retail transactions and Twitter posts." [31], [32]
- Immune system: there is a difference in germline DNA configuration and somatic DNA configuation. The DNA of immunoglobulin genes in lymphocytes (somatic cells) is rearranged in a rather random way. The result is a huge increase of (somatic) information. See my review of Ted Steele's book.
- Chromosome translocation: the famous Philadelphia translocation is a reciprocal translocation between chromosome 9 and 22. The result is that a fusion gene is created by juxtapositioning the Abl1 gene on chromosome 9 to a part of the BCR gene on chromosome 22. The result of the translocation is the oncogenic BCR-ABL gene fusion, which is the cause of leukemia. The point here is that the total genomic information is the same, yet a new gene originated from the translocation. Is this an increase of information or not? [15 Aug 13]
- Meiotic recombination and crossovers: a reciprocal exchange of large pieces between homologous chromosomes. De novo mutations occur. Genetic diversity arises from recombination and de novo mutation, so this is an increase in the information content of the genome [53].
|
- Complexity: currently, there are more than 30 different mathematical descriptions of complexity [17]. What is the most appropriate one for biology? If all these open questions are answered we might end up again with a rather different concept of information in biology [12]. Example: what if two genetic variants in combination cause a specific phenotypic effect which is absent when only one of them is present? Ignore?
- Viruses: A provirus is a virus genome that is integrated into the DNA of a host cell. Example: Retroviruses and Bornaviruses can integrate in the human genome. This increases the information content of the genome. In this way a Bornavirus has created two protein-coding genes in the human genome. Bornaviruses can also integrate into the genome of brain cells (somatic non-heritable integration) [27]. Proviruses may account for approximately 8% of the human genome in the form of inherited endogenous retroviruses.
- Non-coding DNA is DNA which does not contain instructions for making proteins, but nonetheless maybe
transcribed. Recent evidence suggests that some non-coding DNA may be employed by proteins created from coding DNA. (wiki). Non-coding RNAs have a biological function other than coding for protein (wiki). Please note: the very word 'non-coding' and the fact that some have a biological function and thus contain information, but others have unknown function and thus unknown information content. What is the 'real' information content of the genome?
- There is more information in DNA than the primary nucleotide sequence of a genome (that is, the order of A's, C's, G's, and T's). DNA is a molecule with a three-dimensional structure that varies according to the nucleotide sequence. DNA regions that differ on the basis of the order of nucleotides may be similar in structure, which suggests that they may perform similar biological functions (26).
- Biological complexity does not directly correlate with gene number. Daphnia pulex, a crustacean common in lakes and ponds around the world, has 39.000 genes. More than a human (22.000 genes). But we are also probably naïve in defining what is biological complexity. (Science, 5 June 2009)
- Double code. Research over the past few years has revealed that exons not only specify amino acids, they also contain within their sequences cues necessary for intron removal (Scientific American, Jun 2009, p.38). So there is a double code. How do you measure the information content?
- Alternative splicing greatly expands the information content and versatility of the transcriptome through the expression of multiple different mRNAs from individual genes. (Alternative splicing), (see also: double code). In 2018 the total number of protein-coding transcripts (mRNAs) was 267.476 or 12,5 isoforms per protein-coding gene (50). Long non-coding RNA genes (lncRNA) have an average of 2,6 splicing variants. So, it does not make sense to measure the information content of a protein-coding gene on the basis of its sequence when there are many splice variants. But how do you measure it? Summing the length of all the splice-variants? What if the vast majority of observed splice variants correspond to errors?
- Transcriptional noise: "It appears that 95% of the transcribed locations in the human genome are merely transcriptional noise, explained by the nonspecific binding of RNA polymerase to random or very weak binding sites in the genome. We observed over 30 million distinct transcripts in approximately 700,000 distinct genomic locations, of which only about 40,000 (5%) appear to represent functional gene loci." (50). Noise in information processing by humans is per definition ignored because noise is the destruction of information. But in the cell noise is part of reality and cannot be ignored. But how to measure the 'information content'? At the very least the amount of 'noise' should be measured because it is a property of the cell. In fact 30 million distinct transcripts represent information.
- RNA-editing: the information in DNA is not the same as in RNA and proteins because some bases in RNA are replaced by others. See: RNA editing.
- Posttranslational Modification of Proteins: the number of protein modifications can be 2 - 3 orders of magnitude higher than the number of genes in the genomes. This is in large part due to post-translational modifications of proteins that provide covalent alterations to protein backbones and side chains that increase proteome complexities. Greater than 5% of the genes in the human genome encode enzymes that perform such modifications.
- Jean-Jacques Kupiec disagrees with the idea of a deterministic genetic program in development.
Because of the stochastic nature of protein interaction and gene expression, he says, there can be no Aristotelian form or program to give order to life and ward off entropic chaos and death (Nature).
- "Although naked DNA has a relatively static and easy to grasp information capacity, reversible phosphorylation at several sites in even a single protein encodes a potentially large amount of information, and the calculation of this information capacity is complex." (Nature, 8 July 2009)
- Dynamic genome. The assumption that the genome is a static, well-organized library of genes (= biochemical instructions for making cells) is wrong. Cells and genomes cannot be described with the principles of efficient design. Genomes clearly show the imprint of accidents in evolutionary history, selection, and biochemical constraints. Genomes are laden with mechanistic and historical detail; if not always baroque, genomes are clearly not universally elegant in their construction. (source).
- Horizontal Gene Transfer increases the information content of the genome of soma as well of germline cells.
A hefty 8% of human genetic material originates not from intelligent design, neither from our vertebrate ancestors, but from viruses. Bornaviruses are an unforeseen source of genomic innovation [27].
- Information in the brain. There is more information in the brain than in DNA. "How nervous systems can be reconstructed using electron microscopy: neural tissue is cut into slices 40–50 nanometres thick, and then imaged to a resolution of a few nanometres. Imaging 1 cubic millimetre of cortex generates 1 petabyte of data, or about a billion photo images from a typical digital camera." [29]
- Overlapping genes: Multifunctional usage of the same genomic space is common. Overlapping transcripts can be produced from the same or opposite strands of DNA. The regions of overlap of transcripts from opposite strands can include the exons that are present in mature RNAs, or be mostly confined to the introns. (34)
- The extended genome: an estimated 2 to 4 million genes are embedded in the aggregate genome of an intestinal community of ~500 to 1000 bacterial species. The number of distinct viruses in the stool samples ranged from 52 to 2773. The viral sequences are mostly unknown, that is, they have no homologues in the database. Moreover, gut viromes are thought to be highly individual specific as a result of the rapid sequence evolution of phages. This leads to a virome with a vast, uncharted sequence space that is often referred to as biological 'dark matter' [42]. (see also here).
- Paleogenomics: should we include genomes of extinct humans and Hominins such as Neanderthals? The Denisova hominid? See: Paleogenetics.
False positive: Fibonacci pattern found in plants
The Fibonacci series is a sequence of numbers where each number is the sum of the two previous numbers: 1,1,2,3,5,8,13,21,... It was first described by the thirteenth-century Florentine mathematician Leonardo Fibonacci.

Saxifraga Longifolia photo ©Susan Korthof |
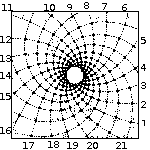
Spirals based on Fibonacci Series
with 13 spirals in one direction and 21 in the other. From: Brian Goodwin |

Brassica oleracea Cultivar Romanesco. © John Walker |
|

sunflower |
A surprising fact is that the Fibonacci series can be found in the arrangement of leaves on the stem of higher plants. In the great majority of plants with spiral arrangement, the arrangement conforms to Fibonacci numbers [6]. Now this looks a perfect case of design [10]. Is it indeed a case of design according to Dembski's Explanatory Filter? Is it a contingent system? The Fibonacci spiral pattern is not the only one present in the plant kingdom. There are other patterns. So there is no necessity. Is it a complex system? It is as complex and as non-random as Dembski's pattern D of binary numbers on page 137. Is it a specified system? A specified pattern needs to be independent of the event. The Fibonacci sequence is independent of the pattern of leaves, because it is 100% determined by the mathematical rules defined by Fibonacci. Is side information involved? Of course: knowledge of the Fibonacci sequence functions as side information and thereby renders the Fibonacci sequence a specification. That side information enables us to construct the Fibonacci pattern to which the leave pattern conforms, without recourse to the actual observation. So we have here Complex Specified Information and so Dembski is forced to conclude intelligent design. But the arrangement of leaves on the stem of a plant is a perfectly natural process, as was shown for example by a simple physical model [6,p115]. So the Fibonacci pattern of leaves is not intelligently caused and is a false positive for Dembski's design criterion. Thereby making the Explanatory Filter an unreliable criterion for design [30].
Dembski's response to my False Positive:
"Korthof fails to appreciate that the design of the biological systems that give rise to Fibonacci sequences is itself in question. Korthof's example is logically equivalent to a computer being programmed to generate Fibonacci sequences. Once programmed, the computer will as a matter of necessity (cf the necessity node of the filter) output Fibonacci sequences." [22,p14].
Remarkably, although the Fibonacci pattern in plants is an excellent example of design according to Demsbski's own criteria, it is not present in his book! Please note that Dembski shifts attention from 'Fibonacci sequences' to 'biological systems that give rise to Fibonacci sequences'. There are two problems with Dembski's answer:
- The Fibonacci pattern is somehow both designed and necessary. But according to his own Explanatory Filter the pattern cannot be designed and necessary at the same time. If it is necessary then it is not designed, and if it is designed it is not necessary. The EF should have eliminated all necessary patterns. That's what the filter is for. What does that "(cf the necessity node of the filter)" mean other than EF eliminated Fibonacci right at the first node? So Fibonacci is not a False Positive but a True Negative? Then he finishes the paragraph with:
"All the computer hardware and software in our ordinary experience is properly referred not to necessity but to design." (NFLp14)
Ordinary experience? OK, but Mr Dembski what does your own Explanatory Filter tell you? It means that EF rejects both the Fibonacci sequence from a computer program and from a biological system. Dembski's way out is to shift from the necessity of the Fibonacci pattern to designed nature of 'biological systems that give rise to Fibonacci sequences'. Now Dembski has the best of both worlds: the Fibonacci pattern is necessary (and fails according to EF) and 'biological systems that give rise to Fibonacci sequences' are designed. But the still unanswered question is why are 'biological systems that give rise to Fibonacci sequences' designed? or in other words:
- Is Dembski's analogy of computer programs and genetic programs a valid analogy? It is just another form of Paley's analogy of watches and organisms. If we assume it is valid, then the designed nature of a computer program is automatically transferred to the design of genetic programs of organisms. But that's begging the question. If Dembski means that anything that produces a Fibonacci sequence is designed, then it is true by definition. It's a tautology. But then Dembski proved nothing at all. Furthermore, for a mathmatician, Dembski does not think and write clearly. It is unintelligible. I have to infer what he means.
|
Circles in the sand
Below is a beautiful picture of circles in fine gravel made by twigs moved by the wind. This natural system has 3 parts: twigs, wind, and fine gravel. Circles are independent specified patterns because they are human invented abstract mathematical objects (side information). The picture demonstrates that blind nonbiological natural forces combined with accidental local conditions are able to create structures that look like Intelligent Design.
 |
| Blowing in the wind: natural circles. ©Susan Korthof |
|
|
| |
Fine Tuning
If everything is designed we don't need a filter. If everything is undesigned we don't need a filter either.
Because some things are designed and some are not, we need a filter. According to Dembski's Explanatory Filter only things not explainable by natural law and/or chance, can be designed. The exclusion of natural law and randomness from the class of designed objects is an essential part of his design inference.
Dembski accepts the Fine Tuning argument (p265). The Fine Tuning argument says that many details of natural laws are designed for life (see Denton, Ross). Dembski accepts the Fine Tuning argument because of improbabilities. However he doesn't make clear that he is talking about improbabilities of natural laws. Aren't these the same natural laws which he excluded in his design inference? For example a snowflake is not designed, but explained by the laws of physics (natural law). But in the end those physical laws are designed by the Designer (according to a theist). So indirectly a snowflake is designed too. But it is contradictory to reject natural law as something that is designed in the design inference, and to accept natural law as something that is designed in the Fine Tuning argument. It undermines the logic of his Explanatory Filter. If everything is designed then the Explanatory Filter has no holes anymore: it catches everything. Please Mr Dembski tell us: are all organisms caught by the filter, or do some (bacteria? viruses?) pass through?
This example highlights a puzzling aspect of Dembski's worldview: the Designer only wrote Complex Specified Information in DNA, but did not design the 4 DNA bases and the genetic code?. He did not design DNA as the carrier of genetic information? This is what fine tuning is all about: ensure that the right materials are there. So a necessary but puzzling assumption of Dembski's Explanatory Filter is that there are un-designed things at all in the universe. Does Dembski really belief that natural laws are not designed?
|
|
| |
The ethical issue: good genes, bad genes
Viruses cause diseases such as smallpox, polio, measles, mumps, rubella, yellow fever, hepatitis-A, influenza, typhus, AIDS. Viruses can create life long damage: for example rubella infection during pregnancy causes damaged eyes, deafness, heart problems, mental retardation of the child. To protect humans against virus diseases, and to help our immune system, vaccines are produced and used all over the world. Viruses are external threats, but there are also enemies within our bodies: oncogenes,
proto-oncogenes (cancergenes such as breast cancer gene) and retroviruses. Viruses also cause tumours (hepatitis-B virus, human papillomavirus, Epstein-Barr virus). There are 'jumping genes' or mobile elements causing havoc in our genome. Borna disease virus (BDV) infects humans, and infects only neurons, establishing a persistent infection in its host's brain. This could explain some psychiatric disorders, such as schizophrenia and mood disorders [27].
Genome size of viruses range from 5000 base pairs or 5 genes (SV40), and 9752 base pairs or 9 genes (HIV), up to 230,000 base pairs or 230 genes (herpesviruses). So all viruses, small and large, far exceed the information content of the cytochrome-c gene and many equal or exceed the CSI of a flagellum (50 genes). So if Dembski claims 'Intelligent Design' for all genes greater than 500 bits, then he cannot deny Intelligent Design for viruses, oncogenes and mobile elements. Regrettably, Dembski comfortably ignored the ethical issue in his book.
|
|
| |
Conclusion
It is clear from the mathematical concepts 'contingency', 'complexity' and 'information' that DNA contains information. But it also follows from the definitions that a piece of 'junk DNA' (noncoding DNA) of 1000 bases has about the same information content as a gene of 1000 bases. This is because both sequences fall into the category of mathematical random sequences. So clearly Dembski needs an extra criterion to detect meaningful DNA and he proposed 'specification'. However a definition of 'specified DNA' is absent in his book. That means that Dembski is not (yet) in a position to make meaningful claims about Complex Specified Information in DNA, let alone claim that a specific piece of DNA is 'intelligently designed'. My main disappointment, however, is that Dembski is not interested in understanding life and evolution.
Notes
- Wen-Hsiung Li (1997) Molecular Evolution, p208-213.
- Nature, 17 June 1999.
- Paul Davies (1999) The Fifth Miracle. The Search for the Origin and Meaning of life.
- "Alchemy, NK Boolean Style" in: Origins & Design 17:2. It's a short review (1600 words) and Dembski started his review with a joke about Kauffman (to set the tone?). With the help of a quote out of context Dembski wants us to believe that Kauffman admits that there is no biological evidence for his theories. But the quoted passage is rather a prelude to biological evidence than Kauffman's admission that no evidence exists, because right after the quote Kauffman deals with biological reality: antibody molecules. Furthermore after the appearance of At Home, evidence has been produced (see: review of At Home) and even a biotech company is now using Kauffman's complex network theory: "The bigger picture", Marina Chicurel, NewScientist 11 Dec 1999 pp39-42.
- Information Content, Compressibility and Meaning by Gert Korthof.
- Brian Goodwin (1995) How the Leopard Changed its Spots, Phoenix Giants. page 109-119, gives a readable description of the Fibonacci series and related phenomena. A rich internet source is: Fibonacci Numbers and Nature. A nice account is given in Philip Ball(2001) The Self-made tapestry. Pattern formation in nature, pp104-109.
- Lee Spetner (1997): Not by Chance!. See review on this site.
- Goodwin (1995) has also nice things to say about this (genetic reductionism!).
- Dean Overman (1997) shows in A Case Against Accident and Self-Organization that Michael Polanyi(1967) in his essay Life's Irreducible Structure was already aware that information in life cannot be reduced to physics and chemistry. See review on this site.
- I later discovered that Dean Overman(1997), p15, actually accepted the presence of the Fibonacci sequence in nature as evidence for intelligent design!
- Hubert Yockey(1992) Information theory and molecular biology, p172. Yockey published these results also in a peer-reviewed scientific journal: "On the information content of cytochrome c", Journal of Theoretical Biology, 1977, 67, 345-76.
- Is there really a law of conservation of information? There are now 2,5 billion websites and 550 billion webpages on the internet and this number increases with 7,3 million websites per day! See: How much information?. Scientific instruments are throwing out increasingly amounts of information. For example, ground-based telescopes in digital sky surveys are currently pouring several hundred terabytes (1012 bytes) of data per year into dozens of archives, enough to keep astronomers busy for decades. And looming on the horizon is the Large Hadron Collider, the world's largest physics experiment, now under construction at CERN, Europe's particle physics lab near Geneva. Soon after it comes online in 2007, each of the five detectors will be
spewing out several petabytes (1015 bytes) of data--about a million DVDs' worth--every year.
Is information as virtual as money? Money can be printed in unlimited quantities. Is there a Law of Conservation of Money? Does the total amount of money on the Earth increase, decrease or is it constant?
- Enrico Coen (1999) The Art of Genes. How organisms make themselves, p310. (review).
- Dembski directly opposes "Information arises from non-information": Manfred Eigen(1996) Steps towards Life, p17.
- Leslie Orgel (1973): The Origins of Life : "Living things are distinguished by their specified complexity." (p189).
- The dystrophin gene is involved in Duchenne muscular dystrophy which is a devastating progressive muscle wasting disorder. The result of it is that the boys go into a wheelchair at about the age of twelve and die in their late teens or early twenties. (Interview with Kay Davies).
- M. Gell-Mann and S. LLoyd (1996) Information Measures, Effective Complexity and Total Information, Complexity, 2, 44-52 (quoted by J. Craig Venter et al in Science 15 Feb 2001).
- Stuart Kauffman (2000) Investigations.
- for example see: Mel Graves(2000) Cancer. The evolutionary legacy, p70-71,77.
- neither in Dembski & Kushiner(2001) Signs of Intelligence. Readers looking for an elaboration of the idea that DNA contains complex specified information will be disappointed: there is nothing. [22-06-01]
- Later Dembski said about this book: "semi-popular work of mine directed toward a theological audience"!!!
- Dembski (2002) No Free Lunch. Rowman & Littlefield Publishers, pp12,14. Also Paperback: 432 pages, Rowman & Littlefield Publishers, Inc.; New edition Feb 2007 432 pp.
- Marc Kirschner & John Gerhart (2005) The Plausibility of Life - Resolving Darwin's dilemma, page 96-104
- Mark Perakh (2004) Unintelligent Design, Prometheus Books, page 130.
- The number of genes in the human genome has steadily decreased: the latest estimate is 20,488 with perhaps 100 more yet to be discovered. Elizabeth Pennisi (2007) Working the (Gene Count) Numbers: Finally, a Firm Answer? Science 25 May 2007: Vol. 316. no. 5828, p. 1113
- Stephen C. J. Parker et al (2009) 'Local DNA Topography Correlates with Functional Noncoding Regions of the Human Genome', Science, 17 apr 2009.
- Cédric Feschotte (2010) 'Virology: Bornavirus enters the genome', Nature 463, 39-40 (7 January 2010) 7 Jan 2010
- Hans Rosling (2011) The Joy of Stats, BBC2 31 Mar 2011.
- Christof Koch (2012) Neuroscience: The connected self, Nature 482, 31 (02 February 2012)
- My example is discussed by Barbara Forrest and Paul R. Gross Creationism's Trojan Horse. The Wedge of Intelligent Design, p.130-131.
- Science 6 April 2012: Vol. 336 no. 6077 p. 22
- "It has been estimated that, from the beginning of civilization – 5,000 years ago or more – until 2003, humanity created a total of five exabytes (billion gigabytes) of information. From 2003 to 2010, we created this amount every two days. By 2013, we will be doing so every ten minutes, exceeding within hours all the information currently contained in all the books ever written." from Nature 26 Apr 2012 in a review of Ignorance: How it Drives Science.
- Joe Gray, Brian Druker (2012) Genomics: The breast cancer landscape, Nature 21 Jun 2012
"Genome-wide measurements of DNA sequence, copy number, structure and gene-expression levels during the past decade have revealed remarkably diverse derangement in individual breast tumours, among different tumours and during various stages of tumour development. These aberrations involve many genes, including several implicated in cancer".
- Philipp Kapranov et al (2007) 'Genome-wide transcription and the implications for genomic organization', Nature Reviews Genetics 8, 413–423 (June 2007)
- Ken A. Dill, Justin L. MacCallum (2012) 'The Protein-Folding Problem, 50 Years On', Science 23 November 2012
- James R. Lupski (2013) 'Genome Mosaicism–One Human, Multiple Genomes', Science 26 July 2013
- Rebecca A. Burrell, et al (2013) The causes and consequences of genetic heterogeneity in cancer evolution, Nature 501, 338–345 (19 September 2013)
- Mel Greaves, Carlo C. Maley (2012) Clonal evolution in cancer, Nature, 19 January 2012: "The inherently Darwinian character of cancer is the primary reason for this therapeutic failure, but it may also hold the key to more effective control". "The evolutionary theory of cancer has survived 35 years of empirical observation and testing, so today it could be considered a bona fide scientific theory." (important review article!)
- January 2014: this problem is still not solved. "William Stafford Noble, a computer scientist at the University of Washington in Seattle, has used deep learning to teach a program to look at a string of amino acids and predict the structure of the resulting protein – whether various portions will form a helix or a loop." Nature, 9 Jan 2014
- A draft map of the human proteome, Nature 29 May 2014.
- Thomas Marcussen et al (2014) Ancient hybridizations among the ancestral genomes of bread wheat, Science, 18 Jul 2014.
- Bas E. Dutilh et al (2014) A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes, Nature Communications (Open)
- Kelly Servick (2014) Harmful mutations can fly under the radar, Science, 19 Sep 2014
- Sleeping dogs of the genome, Science 5 December 2014: "Where does this battle stand today? On one hand, the human retrotransposon load has been reduced to perhaps as few as 100 active elements; yet, two-thirds of the human genome is scarred by the evidence of millions of years of warfare against mobile DNA elements, and new insertions occur at a frequency of 1 per 95 to 270 live births for L1Hs (Long Interspersed Nuclear Element-1 Homo sapiens), and 1 in 20 for Alu."
- Sten Linnarsson (2015) A tree of the human brain, Science, 2 Oct 2015.
- Polyploidy in liver cells, Nature 7 October 2010. Editor's Summary. Original article: "We propose that this mechanism evolved to generate [somatic] genetic diversity and permits adaptation of hepatocytes to xenobiotic or nutritional injury.".
- Gilad D. Evrony (2016) 'One brain, many genomes', Science 04 Nov 2016. "somatic mutations causing neurologic disease "
- Michelle C. Ward, Yoav Gilad (2017) Human genomics: Cracking the regulatory code, Nature, 550, 190-191 (12 October 2017)
- "Pluripotent stem cells show a remarkable ability to self-organize and differentiate in vitro in three-dimensional aggregates, known as organoids or organ spheroids."; "Self-organization implies the formation of ordered structures from relatively homogeneous elements in the absence of an external pattern. In embryology, this involves a dynamic process that starts with a relatively homogenous group of cells that are capable of differentiation and self-patterning and that respond to external forces. The combined action of internal (genetic, biochemical) and external (mechanical) inputs, as well as stochastic events, lead to symmetry breaking, cell rearrangements and non-uniform but controlled spatiotemporal growth.". Sergiu P. Pasca (2018) The rise of three-dimensional human brain cultures, Nature 25 Jan 2018.
- Steven L. Salzberg et all (2018) Thousands of large-scale RNA sequencing experiments yield a comprehensive new human gene list and reveal extensive transcriptional noise, bioRxiv preprint.
- Maria Nattestad et al (2018) Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line, Genome Research.
- Guoliang Chai, Joseph G. Gleeson (2018) A newly discovered mechanism driving neuronal mutations in Alzheimer's disease, Nature News and Views, 21 Nov 2018.
- Bjarni V. Halldorsson et al (2019) Characterizing mutagenic effects of recombination through a sequence-level genetic map, Science, 25 Jan 2019.
- Bennett Childs, Jan van Deursen (2019) Inhibition of 'jumping genes' promotes healthy ageing. Nature, NEWS AND VIEWS 06 February 2019
- Matthew Warren (2019) 'Four new DNA letters double life's alphabet', Nature, 21 Feb 2019. The quote is from Steven Benner. 27 February 2019
- Protein catalogue brings personalized treatments a step closer. Nature 30 April 2019
- Sarah C. Johnson & Sarah E. McClelland (2019) Watching cancer cells evolve through chromosomal instability, NATURE NEWS AND VIEWS 03 June 2019.
- Kristen M. Turner, et al (2017) Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity, Nature 543, 122–125(2017). added: 28 Nov 2019
- Tim H. H. Coorens et al (2019) Embryonal precursors of Wilms tumor, Science 06 Dec 2019 06 Dec 2019
- VISDB: a manually curated database of viral integration sites in the human genome, Nucleic Acids Research, 08 January 2020
- Anders Bergström et al (2020) Insights into human genetic variation and population history from 929 diverse genomes, Science, 20 Mar 2020
- Roxanne Khamsi (2020) Computer algorithms find tumors' molecular weak spots, Science, Jun. 11, 2020
- Pan-cancer analysis of whole genomes, Nature, 5 Feb 2020. Open Access.
- Mapping and characterization of structural variation in 17,795 human genomes, Nature, 27 May 2020. Further: "We report 158,991 ultra-rare structural variants and show that 2% of individuals carry ultra-rare megabase-scale structural variants".
- Elizabeth Pennisi (2020) Controversial DNA modification could play key role in placenta development, Science, Jul. 23 2020.
- 'It will change everything': DeepMind's AI makes gigantic leap in solving protein, Nature 30 Nov 2020.
- Chromothripsis drives the evolution of gene amplification in cancer, Nature,23 December 2020.
Further Reading
 Dembski's 'reply' to my review. (Feedback page) Dembski's 'reply' to my review. (Feedback page)
- Further reading on this site: Stuart Kauffman, Hubert Yockey, Lee Spetner[7], Dean Overman[9], David Foster, Fred Hoyle(1983), Paul Davies[3], Leslie Orgel[15].
- "Intelligent Design as a Theory of Information" by William Dembski, is a summary of chapter 6 of the book reviewed here.
- "Another Way to Detect Design?" by William Dembski, in which he discusses the criticism that Complex Specified Information is not enough to infer intelligent design.
- Abstract of "How Not to Detect Design. Critical Notice: William A. Dembski, The Design Inference", by Brandon Fitelson, Christopher Stephens, and Elliott Sober, Philosophy of Science 66(3) 472-488, September 1999.
- "Intelligent Design is Not Optimal Design" by William Dembski. [ 02-02-2000 ]
- A Dembski resource page maintained by Wesley R. Elsberry.
- Mark Perakh A Consistent Inconsistency -How Dr. Dembski infers intelligent design. "While I am of the opinion that Dembski's effort to create a consistent theory of design failed, I cannot assert that the hypothesis of intelligent design itself is wrong, but only that neither Dembski nor any of his co-believers have so far succeeded in proving it." Please note the beautiful example of the two stones (an irregular and a perfectly spherical shaped ) in the subparagraph "b) Other interpretations of complexity". The pebbles example is described in his book Unintelligent Design on page 130.
- Richard Wein (2002) Not a Free Lunch But a Box of Chocolates A critique of William Dembski's book 'No Free Lunch' [April 23, 2002]
- Brian Charlesworth reviews Dembski's No Free Lunch: Why Specified Complexity Cannot Be Purchased Without Intelligence, Nature 11 July 2002.
- Allen Orr reviews Dembski's No Free Lunch in the Summer 2002 issue of Boston Review. Allen Orr is a well known evolutionary biologist. The review is a must. 26 Aug 2002
- Francis Heylighen (1996) The Growth of Structural and Functional Complexity during Evolution. A very thorough overview. Recommended. Dec 2002.
- Leo P. Kadanoff (2002) "Intelligent Design and Complexity Research. An Essay and Book Review of the Book: No Free Lunch: Why Specified Complexity Cannot be Purchased without Intelligence, William A. Dembski, 429 pp., Roman and Littlefield, 2002". Journal of Statistical Physics, January 2003, Volume 110, Issue 1-2. Kadanoff doubts the relevance of the free lunch theorems to biological systems. Dec 2002
- David Berlinski's reply to the critics in the March issue of Commentary. His original article Has Darwin Met His Match? was published in the Dec 2002 issue of Commentary. He rejects Dembski's design inference. Mar 2003
- Mark Perakh (2003) Unintelligent Design. Hardcover: 420 pages Prometheus Books; (December 2003). Mark Perakh is professor emeritus of physics at California State University, Fullerton. In this book he refutes Intelligent Design theory, especially Dembski. More informaton: Talk Reason (his articles posted there give a reasonably correct image of what there is in the book) or Amazon (reviews and table of contents). Dec 2003
- The Design Revolution is a review of Dembski's latest book by Alistair McBride, minister at Scots Presbyterian Church. Jul 2004
- Matt Young, Taner Edis (2004) Why Intelligent Design Fails. The following chapters discuss William Dembski: Chapter 8: "The Explanatory Filter, Archaeology, and Forensics" (Gary S. Hurd); Chapter 9: "Playing Games with Probability: Dembski's Complex Specified Information" (Jeffrey Shallit & Wesley Elsberry); Chapter 10: "Chance and Necessity-and Intelligent Design?" (Taner Edis); Chapter 11: "There Is a Free Lunch after All: Williarn Dembski's Wrong Answers to Irrelevant Questions" (Mark Perakh). 6 Sep 2004
- William A. Dembski and Michael Ruse, eds., Debating Design: From Darwin to DNA (Cambridge University Press, 2004). (review). 21 Jan 2005
- Thomas D. Schneider 2001 February 9 Evolution of Biological Information The ev model shows explicitly how this information gain comes about from mutation and selection, without any other external influence, thereby completely answering the creationists.
- The Design Revolution: Answering the Toughest Questions about Intelligent Design is a controversial book written by William A. Dembski (2004). (From Wikipedia, the free encyclopedia)
- Peter Olofsson (2007) 'Intelligent design and mathematical statistics: a troubled alliance', Biology and Philosophy, 7 August 2007.
Abstract: The explanatory filter is a proposed method to detect design in nature with the aim of refuting Darwinian evolution. The explanatory filter borrows its logical structure from the theory of statistical hypothesis testing but we argue that, when viewed within this context, the filter runs into serious trouble in any interesting biological application. Although the explanatory filter has been extensively criticized from many angles, we present the first rigorous criticism based on the theory of mathematical statistics.
- Joe Felsenstein (2007) Has Natural Selection Been Refuted? The Arguments of William Dembski, Reports of the National Center for Science Education. "Dembski argues that there are theorems that prevent natural selection from explaining the adaptations that we see. His arguments do not work. Specified information, including complex specified information, can be generated by natural selection without needing to be "smuggled in"."
- S. A. Frank (2008) Natural selection maximizes Fisher information.
- C. Adami (2002) What is complexity? Bioessays (2002) 24:1085-1094.
- John C. Avise (2010) Inside the Human Genome: A Case for Non-Intelligent Design.
|
|

 Mathematician and Intelligent Design Theorist (IDT) William Dembski claims to have established a rigorous criterion for distinguishing intelligently caused objects from natural caused objects. In short: detecting design. He called it the 'explanatory filter'. His previous The Design Inference (1998) can be considered as the mathematical basis of
his latest book Intelligent Design (1999). In this book Dembski gives a popular account of the design inference and explains its relevance to the Creation/Evolution Controversy. In this review I focus on his concept of information discussed in chapters 5 and 6.
The remainder of the book is theology. (21)
Mathematician and Intelligent Design Theorist (IDT) William Dembski claims to have established a rigorous criterion for distinguishing intelligently caused objects from natural caused objects. In short: detecting design. He called it the 'explanatory filter'. His previous The Design Inference (1998) can be considered as the mathematical basis of
his latest book Intelligent Design (1999). In this book Dembski gives a popular account of the design inference and explains its relevance to the Creation/Evolution Controversy. In this review I focus on his concept of information discussed in chapters 5 and 6.
The remainder of the book is theology. (21) Specification 'ensures that the object exhibits a pattern characteristic of intelligence'. Such a pattern cannot be explained by necessity or chance. Dembski's explanatory filter is based on these three concepts. He claims it is a net that catches designed objects. Necessary and random objects pass through. This is Dembski's main claim: specified complexity is a reliable criterion for detecting design. In the natural sciences the design explanation is rejected. Why? What is the problem with Demski's design argument? An obvious target of Dembski's design argument is DNA, because it is claimed to be a contingent, complex and specified string. Let's see where I agree/disagree with Dembski and why:
Specification 'ensures that the object exhibits a pattern characteristic of intelligence'. Such a pattern cannot be explained by necessity or chance. Dembski's explanatory filter is based on these three concepts. He claims it is a net that catches designed objects. Necessary and random objects pass through. This is Dembski's main claim: specified complexity is a reliable criterion for detecting design. In the natural sciences the design explanation is rejected. Why? What is the problem with Demski's design argument? An obvious target of Dembski's design argument is DNA, because it is claimed to be a contingent, complex and specified string. Let's see where I agree/disagree with Dembski and why: